FDA Drug Establishment Registration and FDA Drug Listing
According to U.S. Food and Drug Administration (FDA) drug regulations, all drug establishments that formulate, develop, manufacture, process, or pack drugs intended for marketing in the United States must register with the FDA. Additionally, these establishments must list all their commercially distributed products through the Electronic Drug Registration and Listing System (eDRLS).
This requirement applies to both U.S.-based and foreign manufacturers, processors, packers, and labelers of all drug categories, including Over-the-Counter (OTC), Active Pharmaceutical Ingredients (API), generics, homeopathic products, and prescription drugs.
To complete registration, foreign companies must appoint a U.S. Agent to serve as the FDA’s primary point of contact on their behalf.
A comprehensive list of who must register and list can be accessed on the FDA website.
Importance of a U.S. FDA Agent
The U.S. Agent may be contacted by the FDA for urgent or time-sensitive matters related to the registered drug establishment. Therefore, it is essential that your designated U.S. Agent has a solid understanding of FDA regulations and can effectively communicate FDA requirements to your company.
Moreover, having an independent U.S. Agent—rather than a business partner—can help prevent any potential conflicts of interest.
What PTFP Offers
PTFP (Premium Terra France Pharmaceutical LLC) provides FDA Drug Establishment Registration services for both domestic and foreign companies and can act as your U.S. Agent. We also assist in listing all your marketed products via the FDA’s eDRLS system.
Click the Start button to begin your Drug Establishment Registration and Drug Listing process with PTFP.
FDA Registration Certificate
Once registration is completed, the drug establishment will be issued an FDA registration number. Although the FDA itself does not issue certificates, PTFP—as a third-party agent—can provide a Certificate of FDA Registration for your company.
This certificate serves as proof of FDA registration and can be used for customs clearance, as well as with importers, distributors, and commercial partners. The certificate is valid for one year.
FDA National Drug Code NDC Labeler Code
Under the Drug Listing Act of 1972, all registered drug establishments in the United States are required to submit a current list of all drugs that are manufactured, prepared, propagated, compounded, or processed for commercial distribution to the U.S. Food and Drug Administration (FDA).
Each drug product must be identified and reported using a unique 10-digit, three-segment number called the National Drug Code (NDC). The NDC serves as a universal product identifier for drugs and is made publicly available through the FDA’s NDC Directory.
Structure of the NDC
- Labeler Code (first 5 digits)
- Assigned by the FDA.
- Identifies the manufacturer, packer, or distributor.
- Product Code (next 3–4 digits)
- Assigned by the manufacturer or distributor.
- Identifies the specific strength, dosage form, and formulation of the drug.
- Package Code (last 1–2 digits)
- Also assigned by the manufacturer or distributor.
- Identifies the package size and type.
Note: The full 10-digit NDC format may be displayed in 3-4-3, 5-3-2, or 5-4-1 segment formats depending on the configuration, but will always uniquely identify a specific drug product and packaging.
NDC Listing and Directory
Once assigned and submitted, the NDC and associated drug information are published in the FDA’s National Drug Code Directory, which is accessible to regulatory agencies, healthcare providers, distributors, and the public. This directory plays a critical role in drug tracking, billing, recalls, and regulatory enforcement.
FDA OTC Drug Establishment Registration and OTC Drug Listing
FDA OTC Drugs & OTC Monographs
Over-the-counter (OTC) drug products play an essential role in the U.S. healthcare system. OTC drugs are defined as medications that are safe and effective for use without a prescription from a licensed healthcare provider. There are currently over 300,000 marketed OTC drug products in the U.S.
Rather than reviewing individual OTC drug products, the FDA evaluates therapeutic classes and develops OTC drug monographs—which act as standardized “recipe books.” These monographs specify:
- Permissible active ingredients
- Acceptable dosages
- Approved formulations
- Labeling requirements
Once a monograph is finalized and followed, OTC drug manufacturers can produce and distribute products without FDA pre-approval. The monograph ensures safety, efficacy, and proper labeling standards.
If an OTC product does not conform to an existing monograph, it must go through the New Drug Application (NDA) process for FDA approval. Additionally, cosmetic products making therapeutic claims—such as fluoride toothpastes or anti-dandruff shampoos—must also comply with OTC drug monograph requirements.
OTC Drug Establishment Registration & Listing
Any company (U.S. or foreign) that manufactures, relabels, repacks, or imports OTC drug products for U.S. commercial distribution is required to:
- Register their drug establishment
- List their OTC drug products with the FDA
Registration and listing are conducted via Structured Product Labeling (SPL) format—a standardized XML format adopted by the FDA. The SPL submission must include:
- Establishment details
- Product information
- Drug labels (including Drug Facts Panel and Principal Display Panel)
A complete list of who must register and list can be accessed on the FDA website.
OTC Drug Labeling Requirements
The FDA does not pre-approve OTC drug labels. It is the responsibility of the manufacturer or distributor to ensure their product labeling complies with FDA regulations. Labels must meet strict format and content rules, including:
- Proper Drug Facts Panel layout
- Correct Principal Display Panel presentation
- Disclosure of active/inactive ingredients
- Warnings and directions for use
Failure to comply with these requirements may result in import detentions, market recalls, or enforcement actions.
FDA Registration Certificate
After registration is complete, the FDA assigns the drug establishment an FDA registration number. Although the FDA does not issue a registration certificate, a third-party agent like PTFP can provide you with an FDA Registration Certificate to confirm your registration status. This document is often requested by:
- U.S. Customs
- Distributors
- Importers
- Retailers and other commercial partners
How PTFP Can Help
If you are an OTC drug manufacturer or distributor and need assistance with:
- Identifying the correct OTC monograph
- Completing OTC Drug Establishment Registration
- Submitting SPL format files
- Reviewing OTC product labeling for FDA compliance
Our experts at PTFP are here to support you. Click the Start button or contact us directly for guidance on your OTC drug registration and compliance journey.
FDA Drug Labeling and Ingredient Requirement
All drugs marketed in the United States—regardless of their country of origin—must comply with the Federal Food, Drug, and Cosmetic Act (FDCA). A drug’s intended use is the primary factor that determines which FDA labeling regulations apply to a particular product.
The information permitted on a drug label is closely tied to the drug classification (e.g., prescription drugs, active pharmaceutical ingredients (API), over-the-counter drugs, etc.).
Importantly, the term “labeling” as defined by the FDCA is not limited to the printed label on the outer packaging. It also includes:
- Inserts and inner packaging materials
- Instructional leaflets and usage guides
- Marketing brochures and pamphlets
- Website content or other digital media used for promotion
If a product label includes unauthorized therapeutic claims or content that exceeds the scope of an FDA-approved application or regulation, the drug may be considered misbranded or unapproved, which can trigger enforcement actions or import refusals.
Common Labeling Issues
Labeling violations are among the most common reasons for:
- Delays in FDA approvals
- FDA import alerts
- Customs detentions or product recalls
To avoid such regulatory issues, drug labels must follow specific formatting and content rules, including:
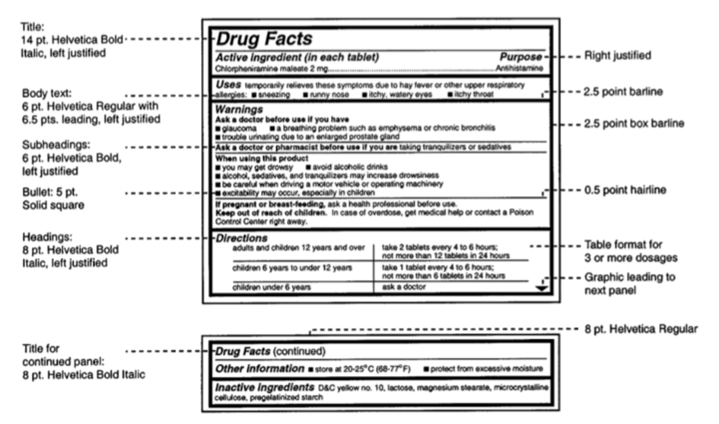
- FDA-compliant drug facts panel
- Proper listing of active and inactive ingredients
- Directions for use, contraindications, and warnings
- Accurate dosage form and strength declarations
- Storage and handling instructions
How PTFP Can Help
Our team of FDA drug labeling experts at PTFP will:
- Review your product label and cross-check it against applicable U.S. federal regulations and databases
- Provide a detailed FDA-compliant labeling report that outlines:
- Required claim modifications
- Suggested graphic layout
- Ingredient declaration adjustments
- Deliver a final labeling recommendation that’s ready for printing and FDA inspection
Contact us to request your free initial FDA labeling assessment and ensure your drug packaging meets all current U.S. regulatory standards.
FDA Drug Color Additives Requirement
All color additives used in drug products must be approved by the U.S. FDA for their specific intended use and formulation. These additives must either:
- Be certified by FDA on a batch-by-batch basis, or
- Be exempt from certification under FDA regulations.
Batch Certification is a regulatory process in which the FDA analyzes the composition of each batch of color additive used in a drug formulation. To obtain this certification, drug manufacturers are required to:
- Submit a sample of the color additive for each production batch
- Provide batch-specific documentation as required by the FDA
Following successful evaluation, the FDA will issue a certificate of approval if the batch sample meets FDA specifications.
Common Compliance Issues
Violations related to unapproved or improperly declared color additives are among the most frequent causes of import refusals and FDA enforcement actions for drug products.
How PTFP Can Help
At PTFP, our regulatory experts support drug manufacturers by:
- Evaluating your current formulation to determine whether your color additives require FDA batch certification or qualify for exemption
- Assisting you in preparing and submitting the appropriate paperwork and color additive samples
- Guiding you step-by-step through the certification and approval process to ensure compliance with FDA requirements
Contact us for assistance with FDA drug color additive review and batch certification.