U.S. FDA Medical Devices Registration and FDA Device Listing
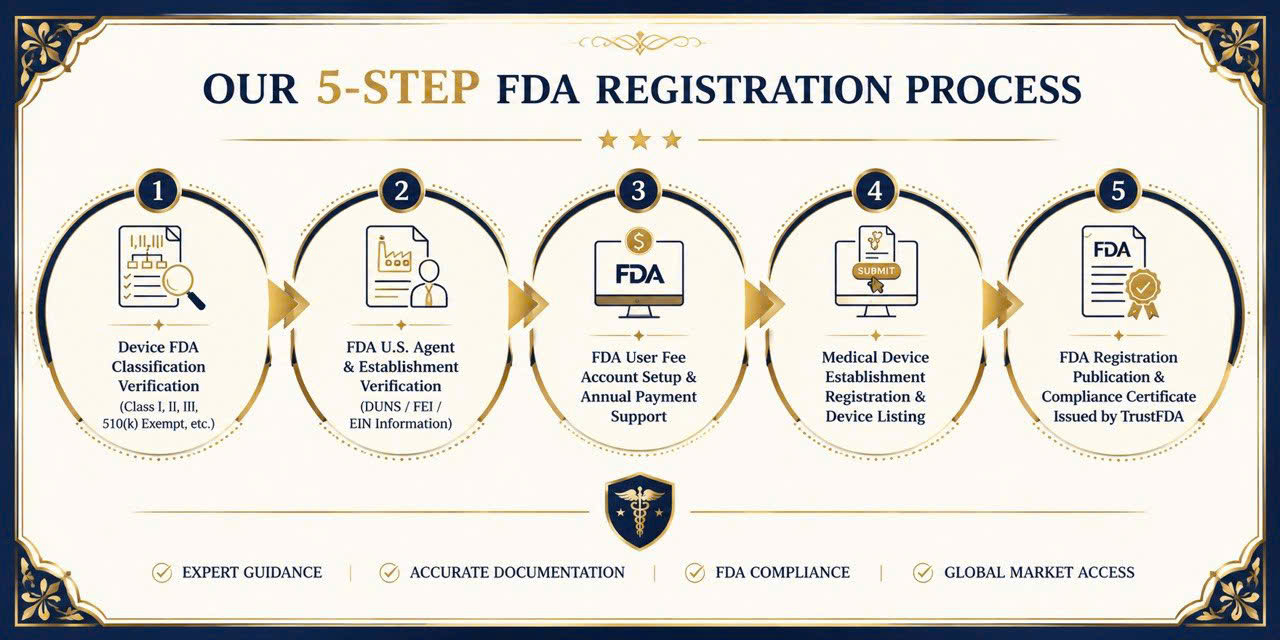
Step-by-Step Registration Process
PTFP provides comprehensive guidance for registering your medical device establishment with the U.S. FDA. The process includes:
- Device classification verification
- U.S. Agent and establishment information review
- Payment account setup
- Device listing procedures
Final registration publication
Who Must Register with the FDA?
According to the U.S. FDA, all medical device establishments that develop, manufacture, assemble, pack, label, export, or import medical devices into the United States must register annually and pay a user fee of $9,280 (for 2025).
In addition, each device must be listed through the FDA’s Device Listing process before entering the U.S. market. No medical device can be legally distributed in the U.S. without a valid device listing number.
Foreign establishments must also appoint a U.S. FDA Agent to act as the official point of contact. Every establishment (domestic or foreign) must assign an official correspondent responsible for managing FDA-related communications.
Establishment registration and device listing must be renewed annually to maintain compliance.
Foreign Exporters and U.S. Importers
Third-party companies that purchase medical devices from foreign suppliers (foreign exporters) are also required to register and pay the FDA annual fee. However, this registration does not exempt the original manufacturer from registering and listing their devices.
Similarly, U.S.-based companies that import medical devices (initial importers) must register and pay the required fee.
What is FDA Device Classification?
Medical devices must be classified correctly as part of the registration process. The FDA categorizes devices into Class I, Class II, or Class III based on risk level, with Class III representing the highest risk.
The FDA Device Classification Database contains the names, descriptions, and product codes that identify each device’s classification.
Once registered, establishment data will appear in the FDA Device Establishment Database. However, device listing numbers remain confidential to protect intellectual property and exclusivity.
Required Information for Registration
To register with the FDA, the following information is required:
- DUNS number and EIN (for U.S. firms)
- Establishment name, address, and official correspondent
- Details of the owner, operator, or agent in charge
- Device classification and list of activities
- Brand or trade names
- U.S. FDA Agent (who must accept the designation)
FDA Registration Steps
- Obtain a DUNS number from the D&B portal or local partner
- Verify establishment and product information
- Submit application to FDA and receive Owner/Operator & Device Listing Numbers
- Receive FDA registration certificate (issued by PTFP as third-party confirmation)
Registration is typically completed within 4–5 business days, provided all necessary information is accurate and available.
Unique Device Identification (UDI)
Since September 2022, manufacturers and labelers of all medical devices (Class I–III) must submit product data to the FDA’s GUDID database and obtain a UDI (Unique Device Identifier) for label compliance.
U.S. FDA Agent Services
Foreign medical device companies are required to appoint a U.S. FDA Agent to register and communicate with FDA. Your U.S. Agent must understand FDA procedures and act promptly in the event of customs holds or regulatory inquiries.
Using an independent U.S. Agent like PTFP helps avoid conflicts of interest and ensures regulatory compliance.
The FDA does not approve or disapprove agent companies but will verify the appointed agent’s agreement to serve during registration, renewal, or updates.
FDA Registration Certificate
While the FDA does not issue certificates, PTFP provides an official Certificate of FDA Registration upon successful registration. This certificate can serve as proof of compliance for:
- U.S. Customs
- Importers and distributors
- Retailers and regulatory authorities
📩 Contact PTFP for expert assistance in registering your medical device establishment and ensuring compliance with all FDA requirements.
FDA Medical Device Labeling Requirements
The U.S. Food and Drug Administration (FDA) maintains strict guidelines regarding the claims and content displayed on medical device labeling. What is allowed on a medical device label is governed by the device’s regulatory classification, including Class I, FDA 510(k) clearance, or FDA Pre-Market Approval (PMA).
Under the Federal Food, Drug, and Cosmetic Act (FDCA), the term “labeling” includes more than just the label affixed to the device. It covers all accompanying materials, such as:
- Instructions for use (IFUs) and manuals
- Marketing brochures and advertising materials
- Product packaging and inserts
- Company websites and digital product listings
Any claim that exceeds the scope of a device’s FDA clearance or approval, or does not conform with the applicable FDA regulation, can result in the product being deemed an unapproved medical device—potentially leading to regulatory enforcement actions, import detentions, or market recalls.
Common FDA Labeling Challenges
Labeling deficiencies are a leading cause of:
- Delays in FDA device clearance or approval
- Rejections or holds at U.S. ports of entry
- FDA warning letters or enforcement actions
How PTFP Can Assist You
At Premium Terra France Pharmaceutical LLC (PTFP), our medical device regulatory team provides specialized FDA-compliant labeling review and advisory services.
Our process includes:
🔹 Comprehensive review of your labeling, claims, and packaging content
🔹 Cross-checking against FDA databases and relevant labeling regulations
🔹 Identification of non-compliant language or claims
🔹 Delivery of a full labeling compliance report
🔹 Suggestions for label design, formatting, and revised claims ready for production
We help ensure that your medical device labeling meets FDA standards before product launch or shipment—saving time, cost, and regulatory risk.
📩 Contact us for a initial labeling assessment and expert FDA labeling review.
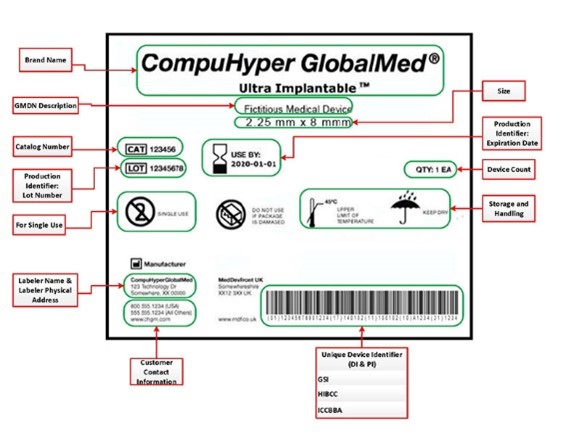
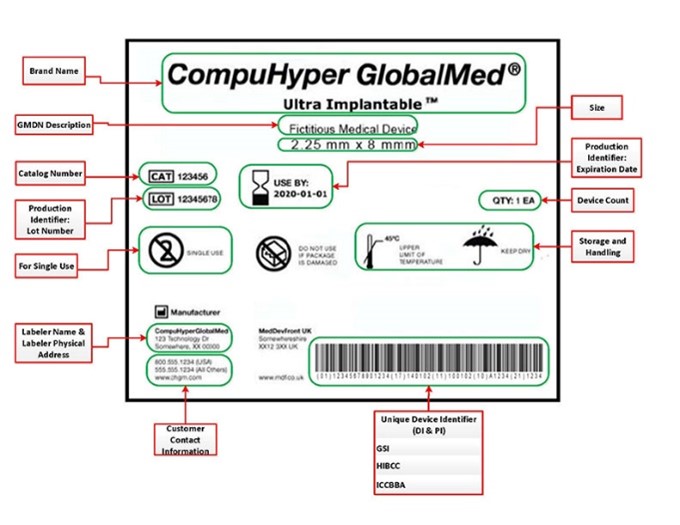
U.S. FDA Global Device Identification GUDID Unique Device Identifiers UDI
The U.S. FDA has implemented a Unique Device Identification (UDI) system designed to improve the identification and traceability of medical devices throughout their distribution and use—especially within hospitals, clinics, and healthcare systems.
Under this system, most medical device labels must include a Unique Device Identifier (UDI) in both human-readable and machine-readable formats (e.g., barcode or QR code).
In addition to labeling requirements, medical device labelers are required to submit device-specific information, including the UDI, to the FDA’s Global Unique Device Identification Database (GUDID). The GUDID is a publicly accessible repository managed by the FDA and serves as a central reference for UDI-related information.
UDI System Objectives
The FDA’s UDI system aims to:
- Enhance patient safety through improved post-market surveillance
- Promote faster identification of devices in case of safety recalls
- Reduce medical errors through precise device tracking
- Streamline supply chain and healthcare IT systems
How PTFP Can Help
Premium Terra France Pharmaceutical LLC (PTFP) offers full support for manufacturers, importers, and labelers of medical devices in navigating UDI compliance. Our services include:
🔹 Determining whether your medical device is subject to UDI requirements
🔹 Preparing and submitting required data to FDA’s GUDID
🔹 Assisting with UDI labeling formats and regulatory requirements
🔹 Providing consultation on label changes to comply with FDA UDI standards
Our regulatory experts will ensure that your UDI data is correctly formatted and submitted, minimizing the risk of errors or delays in FDA processing.
📩 Contact us today for a initial assessment and expert guidance on UDI and GUDID compliance.
FDA 510k Preparations and Submissions
To legally market many medical devices in the United States, manufacturers must obtain FDA clearance through the 510(k) premarket notification process. This requirement applies to most Class II devices, as well as some Class I and Class III devices.
A 510(k) submission is a comprehensive regulatory filing submitted to the U.S. Food and Drug Administration (FDA) to demonstrate that a new medical device is substantially equivalent (SE) in safety and effectiveness to an already legally marketed device, also known as a predicate device.
What Does a 510(k) Submission Involve?
To achieve substantial equivalence, your 510(k) must include:
- A comparison of your device to at least one legally marketed predicate
- Detailed technical and performance data
- Design, material, and intended use comparisons
- Risk assessments, if applicable
- Test reports and clinical data (when required)
The documentation must be formatted according to FDA standards, including administrative, descriptive, performance, and labeling sections.
How PTFP Can Help
At Premium Terra France Pharmaceutical LLC (PTFP), we assist manufacturers and distributors in preparing and submitting complete, FDA-compliant 510(k) submissions.
Our support includes:
🔹 Evaluating device classification and predicate selection
🔹 Assisting in the compilation and formatting of required data
🔹 Reviewing your scientific documentation to ensure FDA readiness
🔹 Preparing the full 510(k) package in accordance with FDA’s Refuse to Accept (RTA) policy
🔹 Submitting the 510(k) on your behalf and assisting with any FDA follow-ups
We help minimize costly delays and improve your chances of successful clearance.
📩 Contact us for expert assistance with your FDA 510(k) submission.
FDA Color Additive Requirements
The U.S. Food and Drug Administration (FDA) strictly regulates the use of color additives in all aspects of the manufacturing process for medical devices. Any color additive used must be explicitly approved by the FDA and accurately declared on the device labeling. The use of unapproved or undeclared color additives is a violation of FDA regulations and may result in enforcement actions.
Batch Certification or FDA Exemption
For color additives used in medical devices, the FDA requires manufacturers to either:
- Obtain batch certification for each lot of the color additive, or
- Secure an exemption from certification under FDA guidelines.
To obtain batch certification, manufacturers must submit a sample of the color additive from each batch along with the necessary documentation to the FDA. If the sample meets all regulatory standards upon examination, the FDA will issue a Certificate of Batch Certification for that specific additive lot.
How PTFP Can Help
Premium Terra France Pharmaceutical LLC (PTFP) supports medical device manufacturers in ensuring that their color additives comply with FDA regulations.
Our services include:
🔹 Initial assessment to determine if FDA batch certification is required
🔹 Guidance in preparing and submitting documentation for batch certification or exemption
🔹 Liaison support during communication with FDA officials
🔹 Regulatory consulting to avoid product detentions or import delays
📩 Contact us today for a free preliminary assessment and professional assistance with your FDA color additive compliance.